Condensed Phase and Interfacial Molecular Science Program
Program Leader
Principal Investigators
The Condensed Phase and Interfacial Molecular Science Program at LBNL supports the DOE Basic Energy Sciences mission to better assess, mitigate and control the efficiency, utilization, and environmental impacts of energy use by providing the molecular basis for understanding chemical, physical, and electron-driven processes in aqueous media and at interfaces (see Figure 1).
Our studies seek breakthroughs in the fundamental understanding of electron and molecular dynamics at interfaces, solvation in electrolyte solutions and at interfaces, and chemical reactions and transformations in systems often far from equilibrium. These projects are unified by a long term vision of understanding how rare events and fluctuations govern chemical reactivity in heterogeneous environments.
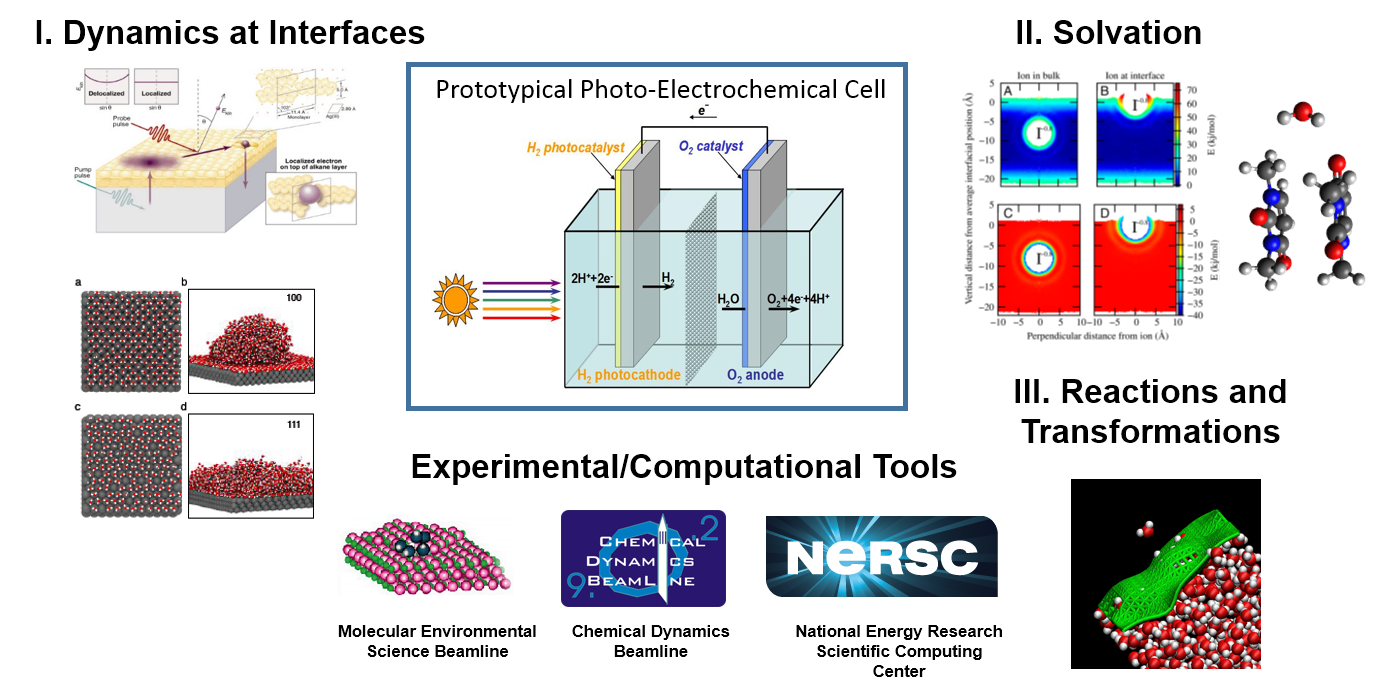 |
| Figure 1: The long term objective of our work is to expand the fundamental science base that underlies current and future problems in energy sciences, which include both energy production and its environmental impacts. Our three projects (described below) target three of the many processes that underlie current and future energy sciences illustrated by a current example: a prototypical photo-electrochemical cell. Our work relies on unique computational and experimental resource at Berkeley Lab. |
The Charge and Molecular Dynamics at Interfaces project investigates charge transfer steps in catalytic reactions, interfacial electron relaxation, localization and transport across metal/adsorbate interfaces and theoretical descriptions of water dynamics at solid interfaces.
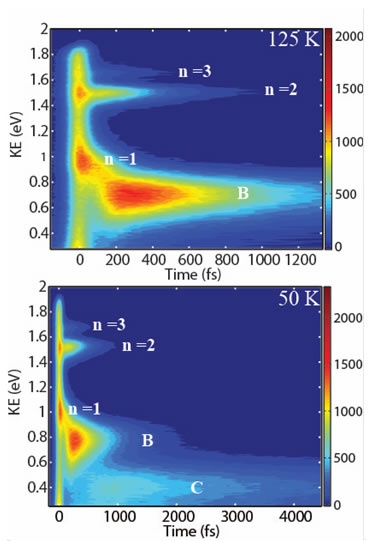 |
| Figure 2. TPPE spectrum ultrathin films of NaCl on Ag(100). Top: Spectrum of sample at 125 K. Bottom: Spectrum of sample at 50 K. n=1,2, and 3 correspond to the delocalized IPS, B are electrons trapped at low coordinated sites, and C is the new electronic state seen at low temperature. |
Time resolved spectroscopy is used to understand how molecules influence charge dynamics at solid interfaces (see Figure 2) and at catalytic surfaces. Theory elucidates the dynamics of water and other liquids at solid-liquid interfaces, using coarse grained models to treat universal features and molecular dynamics simulations with force fields developed by electronic structure calculations to treat atomistic details. Our research goal is to determine how molecular dynamics couple with charge dynamics at interfaces. A range of transient spectroscopic techniques are employed to follow the transfer of charges leaving the solid side, the rise and decay of intermediate molecular species from the liquid/adsorbate side, and to probe the evolution of purely interfacial electronic states.
Our Solvation project explores the solvation properties of ions and molecules which play critical roles in electrochemical transport, cloud droplet formation, ion pairing, crystallization, protein folding, corrosion, as well as chemical reactions in solutions and at interfaces.
We are investigating the solvation of electrons at metal interfaces, solutes in bulk liquids and at interfaces and within small clusters. Here theory elucidates experiment through both the construction of coarse-grain models of solvation (see Figure 3) in heterogeneous environments and by first principles calculations of electronic structure. We use nonlinear spectroscopy to measure selected ion absorption at aqueous interfaces, and develop generalized theories of solvation in heterogeneous environments using atomistic and coarse grained simulations.
We seek to develop predictive models of both bulk electrolyte behavior and interfacial phenomena that are essential for future advances in energy research. This subtask addresses the development and application of novel spectroscopic (X-ray absorption, VUV photoionization, nonlinear optics and two photon photoemission) probes of electron solvation in ionic liquids at metal interfaces, contact ion pairing, bulk solvation structure of Li+ ions, interfacial solvation of simple solutes, and micro-solvation in clusters. Theory and simulation establish the basic principles that govern liquid water accommodation of molecular solutes. Future directions include the solvation properties of free radicals, size-selected clusters, and the development of a general field theory of solvation.
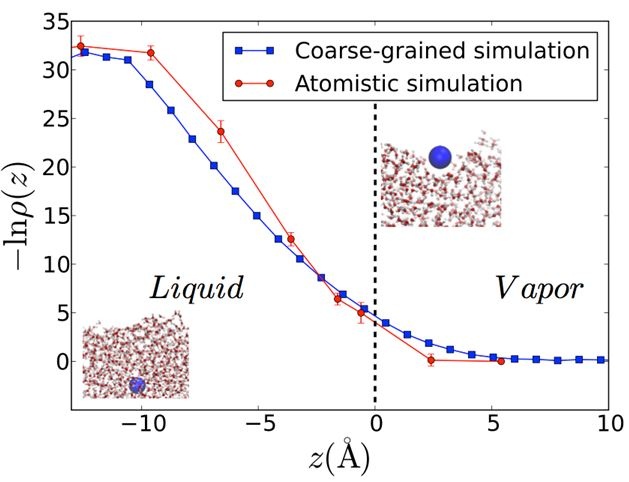 |
| Figure 3: Free energy profile of a hydrophobic solute (5Å in diameter) crossing through the liquid-vapor interface. The influence of capillary fluctuations is strongly evident from variations of the average solute density ρ(z) at heights z more than twice the solute diameter away from the average position of the interface (z=0). |
The Reactions and Transformations project addresses multiphase chemistry, bulk phase dissolution, catalytic reactions, self-assembly dynamics, and transformations at interfaces, glasses and in confined water. This work confronts the complexity of bond breaking and making and seeks to clarify the role of solvent fluctuations and rare events in chemical and multi-phase transformations.
This research aims to establish the chemical methods and insight that will enable understanding of systems that are profoundly complex in composition, heterogeneity, preparation, and organization. The case studies span a broad range: from bulk phase and interfacial chemical reactions to the chemistry of self-assembly. Long-term goal: transitioning from the chemical physics of precisely specified model systems to transformations within the disordered and incompletely controlled environments that feature in energy applications. Efforts to understand how the transformations depend upon chemical heterogeneity, spatial confinement, the slow dynamics of vitrification and coarsening, and the interplay between surface and bulk dynamics. Mass spectrometry is used for characterization of strongly heterogeneous samples with proposed work to examine transformations at complex interfaces under ambient conditions. New theoretical studies of the dynamics of self-assembly are envisioned, whose microscopic description involves extending simulation tools to examine fluctuations on disparate time and length scales.
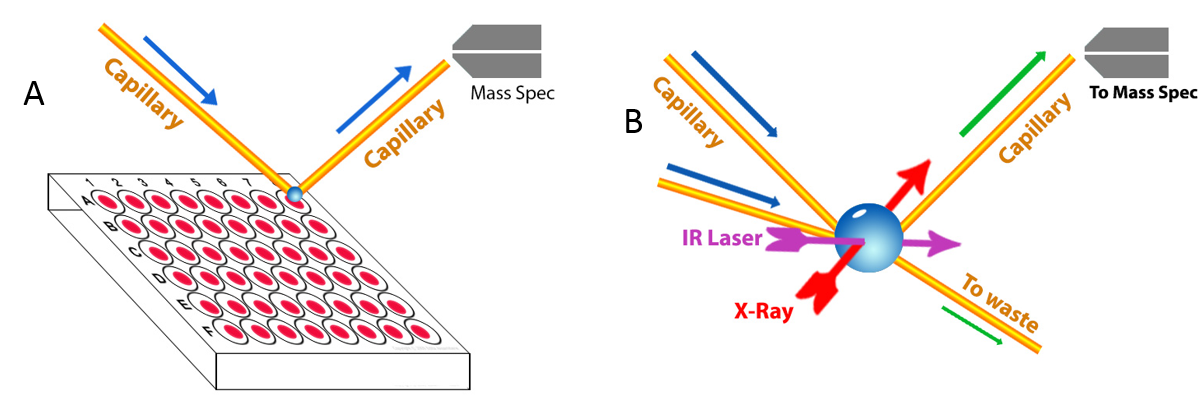 |
| Figure 4 (A) NanoDESI on microfluidic reactors, (B) Droplet reactor |
Work in all of these projects requires the continual development of new experimental and theoretical tools. A key feature and challenge in describing any complex system is the presence of multiple time and length scales that together ultimately govern average chemical transformations. We are developing microfluidic devices (see Figure 4) to study transient intermediates produced in chemical reactions using X-ray spectroscopy, and are making new measurements of VUV and X-ray spectra of size-selected clusters. Microsolvation in clusters not only enables stringent tests of quantum chemistry but is often used as a simplified model of bulk liquids such as water or methanol.