
The Chemical Sciences Division is Berkeley Lab’s home for fundamental research in chemistry and chemical engineering. Our work provides the starting points for new and advanced energy technologies and for mitigating the environmental impacts of energy use.

The CSD core research programs address fundamental questions in chemical sciences that are central to the mission of the Department of Energy’s Office of Science. Topics include catalysts and catalytic reactions; transformations at interfaces; chemical conversion processes associated with energy production; the chemistry of actinides and transactinides; and in-depth investigations of chemical processes.

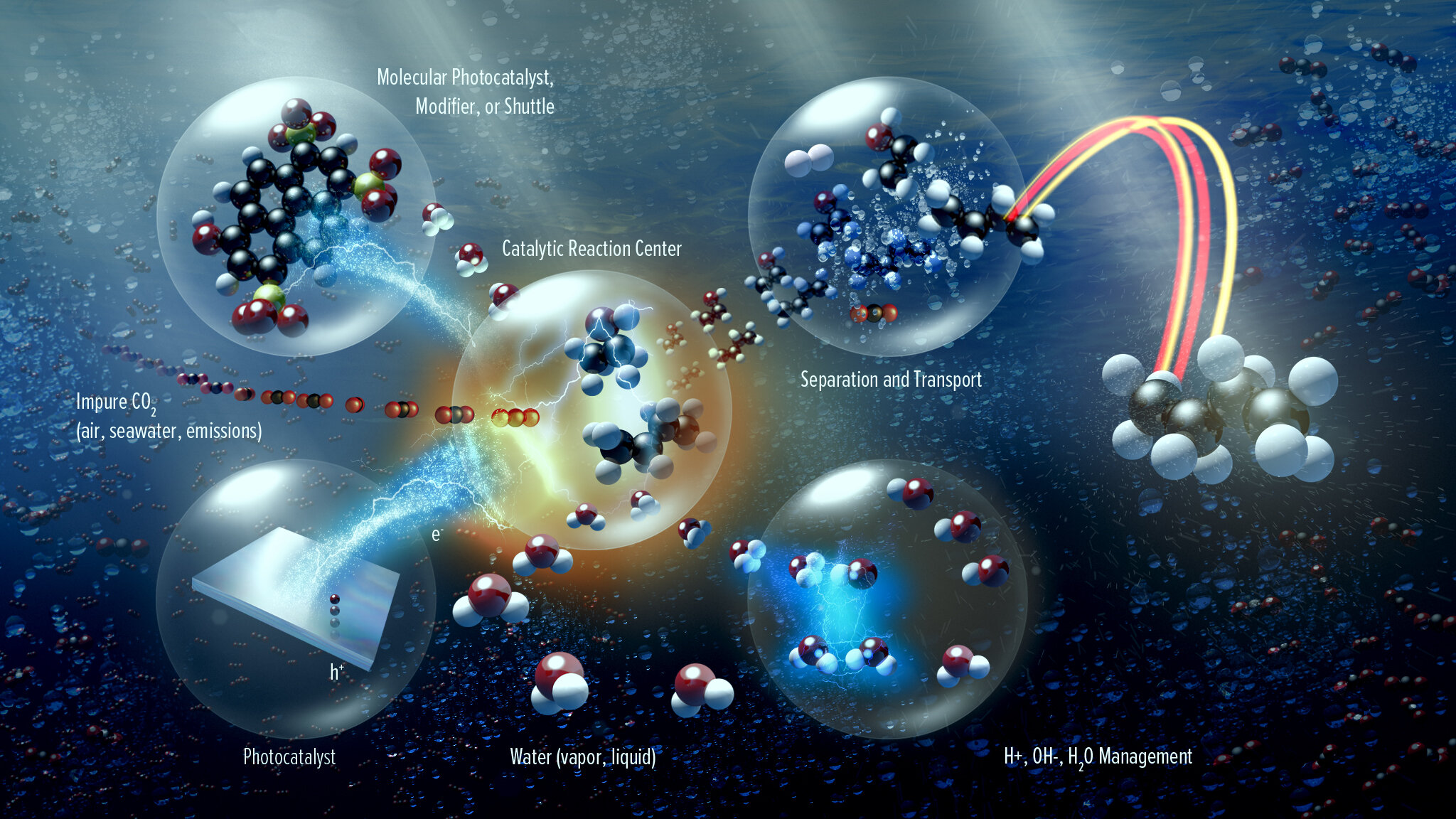
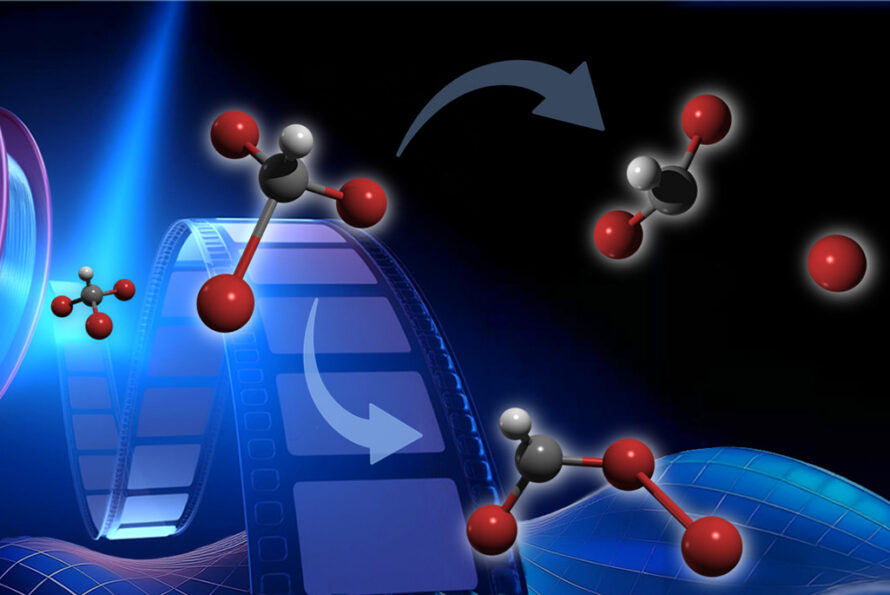
In addition to the core scientific programs, CSD researchers work to develop new science and the underpinning basic knowledge for future energy technologies and solutions. Current projects include new and better methods for the separations of critical elements, chemical re- and upcycling of polymers, the design of molecular qubits, the light- and electron-driven conversion of carbon dioxide to liquid fuels, and the development of computational methods to explain and then predict new chemical reactions.
We have built, and now operate, some unique facilities to enable us to characterize the compounds and catalysts that we make, and to manipulate some of the most challenging isotopes in the periodic table, such as the transuranic elements.



An integrated, state-of-the-art cluster of research buildings for materials and chemistry sciences is envisioned to be built at the top of Charter Hill adjacent to the Advanced Light Source. The vision for the Charter Hill Campus is to support multidisciplinary research that bridges basic and applied science.
